
Proc.
Nati.
Acad.
Sci.
USA
Vol.
87,
pp.
3147-3150,
April
1990
Biochemistry
Mutation
eliminating
mitochondrial
leader
sequence
of
methylmalonyl-CoA
mutase
causes
mut0
methylmalonic
acidemia
(molecular
cloning/enzyme
precursors/signal
peptides/inborn
error
of
metabolism/amino
acid
sequence)
FRED
D.
LEDLEY*t4§,
RUUD
JANSEN*,
SANG-UK
NHAM*,
WAYNE
A.
FENTON¶,
AND
LEON
E.
ROSENBERG¶
*Howard
Hughes
Medical
Institute,
and
Departments
of
tCell
Biology
and
§Pediatrics,
Baylor
College
of
Medicine,
Houston,
TX
77030;
and
IDepartment
of
Human
Genetics,
Yale
University
School
of
Medicine,
New
Haven,
CT
06510
Contributed
by
Leon
E.
Rosenberg,
January
8,
1990
ABSTRACT
Methylmalonyl-CoA
mutase
(EC
5.4.99.2)
is
a
mitochondrial
matrix
enzyme
whose
activity
is
deficient
in
the
inherited
disorder
methylmalonic
acidemia.
Previous
studies
on
primary
fibroblast
cell
lines
from
patients
with
methyl-
malonic
acidemia
have
delineated
a
variety
of
biochemical
phenotypes
underlying
this
disorder.
One
cell
line
with
primary
mutase
apoenzyme
deficiency
exhibited
a
particularly
unusual
phenotype;
it
expressed
an
abnormally
small
and
unstable
immunoreactive
protein,
which
was
not
imported
by
mitochon-
dria.
We
now
report
cloning
and
sequencing
of
the
cDNA
encoding
this
mutant
protein.
The
mutation
is
a
single
base
change,
a
cytosine
--
thymine
transition,
which
introduces
an
amber
termination
codon
at
position
17
within
the
mitochon-
drial
leader
sequence.
The
immunoreactive
protein
produced
by
these
cells
reflects
translation
from
AUG
codons
down-
stream
from
this
termination
codon
and,
hence,
lacks
a
mito-
chondrial
leader
peptide.
This
mutation
represents
a
complex
prototype
for
a
class
of
mutations
in
which
absence
of
the
mitochondrial
targeting
sequence
leads
to
absence
of
a
func-
tioning
gene
product.
Methylmalonyl-CoA
mutase
[MCM;
(R)-2-methyl-3-oxopro-
panoyl-CoA
CoA-carbonyl
mutase,
EC
5.4.99.2]
is
a
mito-
chondrial
matrix
enzyme
encoded
by
a
nuclear
gene
(1-3).
It
is
translated
on
cytoplasmic
ribosomes
as
a
742-amino
acid
(82,145
Da)
precursor
protein
comprising
a
32-amino
acid
mitochondrial
leader
sequence
and
a
710-amino
acid
(78,351
Da)
mature
apoenzyme
(4).
The
leader
sequence
directs
the
precursor
to
the
mitochondria
where
it
is
recognized,
trans-
located
across
both
mitochondrial
membranes,
and
cleaved
by
a
specific
matrix
endoprotease
to
form
the
mature
subunit
(3).
The
subunits
then
assemble
into
a
homodimer
and
bind
the
adenosylcobalamin
cofactor.
Inherited
deficiency
of
MCM
causes
methylmalonic
aci-
demia
(MMA),
an
inborn
error
of
organic
acid
metabolism
in
which
failure
of
methylmalonyl-CoA
degradation
leads
to
widespread
aberrations
in
organic
acid,
amino
acid,
and
carbohydrate
metabolism
(5).
MMA
can
result
from
two
general
classes
of
genetic
defects:
those
termed
cbl,
which
involve
genes
required
for
provision
of
the
adenosylcobal-
amin
cofactor
(McKusick
nos.
251100
and
151110)
(5,
6),
and
those
termed
mut,
which
involve
the
gene
encoding
the
MCM
apoenzyme
(McKusick
no.
251000)
(5,
6).
mut
MMA
is
further
divided
into
phenotypic
subgroups
based
on
the
presence
(mut
-)
or
complete
absence
(muto)
of
detectable
enzyme
activity
in
primary
fibroblasts
(5,
7).
The
mut0
phenotype
itself
is
pleomorphic,
with
some
cells
containing
significant
amounts
of
cross-reactive
antigenic
material
(CRM),
some
expressing
severely
decreased
CRM,
some
expressing
unstable
proteins,
which
can
only
be
detected
by
pulse-chase
or
in
vitro
translation
methods
(8,
9),
and
some
expressing
no
detectable
CRM.
More
recent
studies
using
the
cloned
MCM
cDNA
have
shown
that
some
mut0
cells
have
extremely
low
levels
of
MCM
mRNA,
whereas
other
muto
and
all
mut
-
cells
have
mRNA
that
is
grossly
normal
in
quantity
and
size
(10,
11).
One
particularly
interesting
mut0
cell
line
(FB552)
has
been
described
in
which
the
mutant
MCM
gene
expresses
a
protein
that
is
smaller
than
the
normal
precursor
and
is
highly
unstable.
This
abnormal
protein
is
not
imported
or
cleaved
by
mitochondria
(9).
This
cell
line
exhibits
no
detectable
enzy-
matic
activity
and
no
stable
CRM
in
culture
(8,
9),
despite
having
grossly
normal
hybridizing
MCM
mRNA
(11).
These
data
suggest
that
the
mutant
gene
product
is
deficient
in
the
mitochondrial
targeting
signal
(9).
We
now
report
cloning
and
sequencing
of
the
MCM
cDNA
from
the
FB552
cell
line
and
identification
of
the
mutation
that
gives
rise
to
this
pheno-
type.
MATERIALS
AND
METHODS
Cloning
of
MCM
cDNA
by
the
Polymerase
Chain
Reaction
(PCR)
and
Determination
of
the
Nucleic
Acid
Sequence.
MCM
cDNA
was
cloned
by
reverse
transcription
and
PCR
ampli-
fication
from
total
RNA
by
using
the
methods
illustrated
in
Fig.
1A
(12).
Briefly,
RNA
was
prepared
by
the
hot
phenol
method
(13),
and
first
strand
cDNA
was
prepared
by
priming
reverse
transcription
with
an
oligonucleotide
complementary
to
sequences
in
the
3'
untranslated
region
of
MCM
(14).
Two
overlapping
portions
of
the
cDNA
were
amplified
by
using
oligonucleotides
corresponding
to
the
MCM
cDNA
se-
quence, subcloned
directionally
in
pGEM7zf(+),
and
se-
quenced
by
dideoxy
sequencing
by
using
oligonucleotides
complementary
to
vector
or
cDNA
sequences
as
illustrated
in
Fig.
1B.
PCR
mistakes
and
heterozygosity
were
evaluated
by
sequencing
single
isolates
of
cloned
material
and
pools
containing
15-20
independent
isolates.
Recombinant
DNA
procedures
were
carried
out
by
using
standard
methods
(14).
Oligonucleotide
sequences
will
be
given
elsewhere.
In
Vitro
Transcription
and
Translation
of
MCM
cDNA.
The
full-length
normal
and
mutant
cDNA
sequences
were
recon-
structed
by
three-part
ligation
of
the
5'
Cla
I-Acc
I
and
3'
Acc
I-Mlu
I
fragments
into
a
Cla
I-Mlu
I-digested
pGEM7zf
vector.
Subclones
of
the
full-length
MCM
cDNA
containing
5'
terminal
exonuclease
III
deletions
have
been
described
(4)
(see
Fig.
3A).
These
include
hMCM26
(bases
315-2770),
hMCM1.
1
(bases
424-2770),
hMCM5.1
(bases
721-2770),
and
hMCM8.3
(bases
928-2770).
Synthetic
RNA
was
produced
by
transcription
from
the
T7
promoter
by
T7
RNA
polymer-
ase
(Promega)
and
was
translated
into
protein
by
using
reticulocyte
lysate
(Amersham).
Incorporation
of
[35S]methi-
Abbreviations:
MCM,
methylmalonyl-CoA
mutase;
MMA,
methyl-
malonic
acidemia;
PCR,
polymerase
chain
reaction;
CRM,
cross-
reactive
antigenic
material.
*To
whom
reprint
requests
should
be
addressed.
3147
The
publication
costs
of
this
article
were
defrayed
in
part
by
page
charge
payment.
This
article
must
therefore
be
hereby
marked
"advertisement"
in
accordance
with
18
U.S.C.
§1734
solely
to
indicate
this
fact.

Proc.
Natl.
Acad.
Sci.
USA
87
(1990)
A
Clal
Accl
Kpnl
BcIl
AAAAA
*-
33
53
3
28
2
C
a
ll
pna
pGEM7zf(+)
pGEM7zf
(+)
B
5P6-.
SP
...
...
2
48
-
T7
NORMAL
cDNA
MUTANT
cDNA
SiNGLE
POOLED
IA
T
C
G
A
T
C
G
A
T
C
Gc
=
S.
t~
i~~~
GLN
...
.ACCTGAGGCAG...
...ACCTGAGGTAG
...
TOP
READING
FRAME
t
t
C(128)
HindlIl(-)
BASES
AAAAA
cDNA
SEQUENCE
1000
2000
3000
T7
-
34
-
35--
16
-
-*28
-*37
-5SP6
-*
15
-
17
BASES
1000
2UUU
FIG.
1.
Strategy
for
cloning
and
sequencing
cDNA
from
FB552.
(A)
Schematic
showing
oligonucleotides
used
for
reverse
transcrip-
tion
and
PCR
amplification
of
FB552
cDNA.
The
restriction
sites
used
for
directional
cloning
of
PCR
products
are
shown.
(B)
Strategy
for
sequencing
FB552
cDNA
showing
the
positions
of
oligonucleo-
tide
primers
internal
to
the
cDNA
sequence
and
the
direction
of
sequencing.
The
full-length
cDNA
is
described
in
ref.
4.
The
se-
quences
of
the
numbered
oligonucleotides
will
be
given
elsewhere.
onine
(Amersham)
into
protein
was
assayed
on
SDS/7-15%
polyacrylamide
gradient
gels.
Demonstration
of
Mutation
in
Genomic
DNA
by
PCR.
Genomic
sequences
spanning
the
putative
mutation
site
in
exon
II
and
a
previously
described
HindIII
polymorphism
in
exon
III
were
amplified
by
PCR
using
oligonucleotides
com-
plementary
to
exon
and
intron
sequences
(S.-U.N.,
R.J.,
and
F.D.L.,
unpublished
data)
(see
Fig.
4A).
The
presence
of
the
mutation
was
confirmed
by
direct
sequencing
of
PCR
prod-
ucts
using
methods
described
(16,
17).
The
HindIII
polymor-
phism
was
identified
by
digestion
of
PCR
products,
separa-
tion
of
fragments
on
a
15%
acrylamide
gel,
and
staining
with
ethidium
bromide
(1
gg/ml)
(S.-U.N.,
R.J.,
and
F.D.L.,
unpublished
data).
In
the
direct
analysis
of
PCR
products
by
sequencing
or
restriction
digestion,
PCR
mistakes
represent
minor
species,
which
are
not
detectable.
RESULTS
Sequence
of
the
FB552
cDNA.
Several
differences
from
the
published
reference
MCM
sequence
(hMCM7.1)
(4)
were
observed
in
the
cDNA
cloned
from
FB552.
The
mutation
identified
in
the
FB552
cDNA
is
a
transition
of
C128
-T128
which
changes
the
CAG
codon
encoding
Gln-17
to
a
TAG
(amber)
termination
codon.
There
was
no
evidence
of
the
C128
base
in
the
pooled
sample,
indicating
that
all
of
the
mRNA
contained
the
T128
variant
(Fig.
2).
We
also
observed
a
previously
described
HindIII
polymorphism
at
base
712.
Again,
there
was
no
evidence
for
heterogeneity
in
the
mRNA;
all
of
the
cDNAs
exhibited
the
HindIII(-)
sequence.
FIG.
2.
Mutation
in
FB552
cDNA.
The
sequencing
gel
shows
the
sequence
of
a
normal
cDNA
clone,
an
isolated
single
mutant
cDNA
clone,
and
a
pool
of
15
mutant
cDNA
clones.
The
C128
-T128
transition
is
apparent
when
comparing
the
normal
and
isolated
clone
sequences.
The
pooled
sequence
shows
only
T128,
indicating
that
all
of
the
mRNA
contains
this
mutation.
This
mutation
introduces
an
amber
termination
codon
at
position
17
within
the
mitochondrial
leader
sequence.
The
position
of
the
mitochondrial
leader
sequence
(open
box),
mature
apoenzyme
sequence
(solid
box),
and
cDNA
3000
sequence
are
shown
schematically
along
with
the
position
of
the
mutation
and
HindI11
polymorphic
site.
In
Vitro
Transcription
and
Translation
of
cDNA
Bearing
the
FB552
Mutation.
The
5'
segment
of
the
FB552
cDNA,
comprising
bases
52-375,
was
recombined
with
a
normal
3'
cDNA
segment
in
the
vector
pGEM7zf(+).
RNA
was
tran-
scribed
from
this
clone,
from
a
control
plasmid
containing
the
normal
52-2774
sequence,
and
from
a
series
of
clones
con-
taining
5'
terminal
deletions
(Fig.
3A).
These
clones
were
selected
such
that
the
first
AUG
of
each
clone
represents
a
different
internal
AUG
(methionine)
codon
and
potential
initiation
site
(Fig.
3A).
In
vitro
translation
of
the
normal
sequence
produces
the
expected
precursor
protein
of
-82
kDa
as
well
as
a
series
of
smaller
translation
products
(Fig.
3B,
lane
3).
The
recon-
structed
mutant
clone
does
not
exhibit
the
82-kDa
peptide,
as
expected,
although
all
of
the
smaller
translation
products
are
present
(Fig.
3B,
lane
2).
The
terminally
deleted
clones
exhibit
a
sequential
loss
of
the
larger
molecular
mass
trans-
lation
products
(Fig.
3B,
lanes
5-8)
corresponding
to
initia-
tion
at
AUG
codons
within
the
cDNA,
which
can
be
indi-
vidually
identified
by
reference
to
the
terminally
deleted
subclones
(Fig.
3A).
Specifically,
clone
hMCM26
lacks
the
first
internal
methionine
codon
at
position
311
and
does
not
make
either
the
82-kDa
protein
or
the
70-kDa
protein.
Clone
hMCM1.1
(Fig.
3B,
lane
5)
lacks
the
second
internal
methio-
nine
codon
at
position
371
and
lacks
the
63.9-kDa
translation
product.
The
dominant
band
of
material
translated
from
FB552
corresponds
to
the
product
of
-45
kDa
translated
from
clone
hMCM8.3
(Fig.
3B,
lane
7).
It
is
not
possible
to
correlate
all
of
the
bands
observed
by
in
vitro
translation
of
the
recombinant
RNA
with
those
observed
from
purified
cellular
mRNA,
because
plasmid
and
polylinker
sequences
at
the
5'
end
of
the
recombinant
RNA
transcript
may
affect
translational
efficiency
(18).
There
are
no
other
reading
frames
in
the
MCM
cDNA
that
would
encode
proteins
>10
kDa.
Identification
of
Mutation
and
Polymorphisms
in
FB552
Genomic
DNA.
To
confirm
the
presence
of
the
C128
->
T128
mutation
in
the
FB552
genome,
DNA
from
the
FB552
cell
line
was
amplified
by
PCR
and
subjected
to
direct
sequencing
by
3148
Biochemistry:
Ledley
et
al.
Proc.
Natl.
Acad.
Sci.
USA
87
(1990)
3149
A
LANE
V
w
WWv
wV
w
3
v
4
t
v
5
v
6
v
7
v
CLONE
LARGEST
PRODUCT
MCM7.1
82,145
MCM26
70,801
MCM
1.1
63,942
MCM5.1
56,751
MCM8.3
47,046
A2/
HlndIII(+/-)
t
EXON
II
,'~~N
II
#64
-0
'O
#47
88bp1168bp
#23
4
*
#59
B
A
T
C
G
1000
2000
3000
BASES
B
1
2
3
4
5
6
7
8
..
rs
A
CT
1
2
8
-=
-
-
z
_
t.
RDo
w~~~9
7
_
w
66.3
FIG.
3.
In
vitro
transcription
and
translation
of
cDNA
with
the
FB552
mutation
and
of
controls.
(A)
Schematic
of
potential
initiation
sites
within
the
long
open
reading
frame
of
the
MCM
mRNA
and
the
terminally
deleted
subclones
used
for
in
vitro
transcription
and
translation.
v,
Position
of
"in-frame"
methionine
(AUG)
codons;
A,
position
of
the
normal
termination
codons;
I
,
position
of
the
amber
mutation
(position
128).
No
alternative
reading
frames
that
could
code
for
translation
products
>10
kDa
are
present.
(B)
In
vitro
translation
of
clones
containing
normal
MCM
cDNA,
of
cDNA
carrying
the
FB552
mutation,
and
of
deletion
subclones.
Lane
1,
no
RNA;
lane
2,
translation
of
mRNA
carrying
FB552
mutation;
lane
3,
translation
of
normal
MCM
mRNA;
lane
4,
translation
of
mRNA
from
clone
hMCM26;
lane
5,
translation
of
mRNA
from
clone
hMCM1.1;
lane
6,
translation
of
mRNA
from
clone
hMCM5.1;
lane
7,
translation
of
mRNA
from
clone
hMCM8.3;
lane
8,
molecular
size
markers
(sizes
in
kDa
are
indicated).
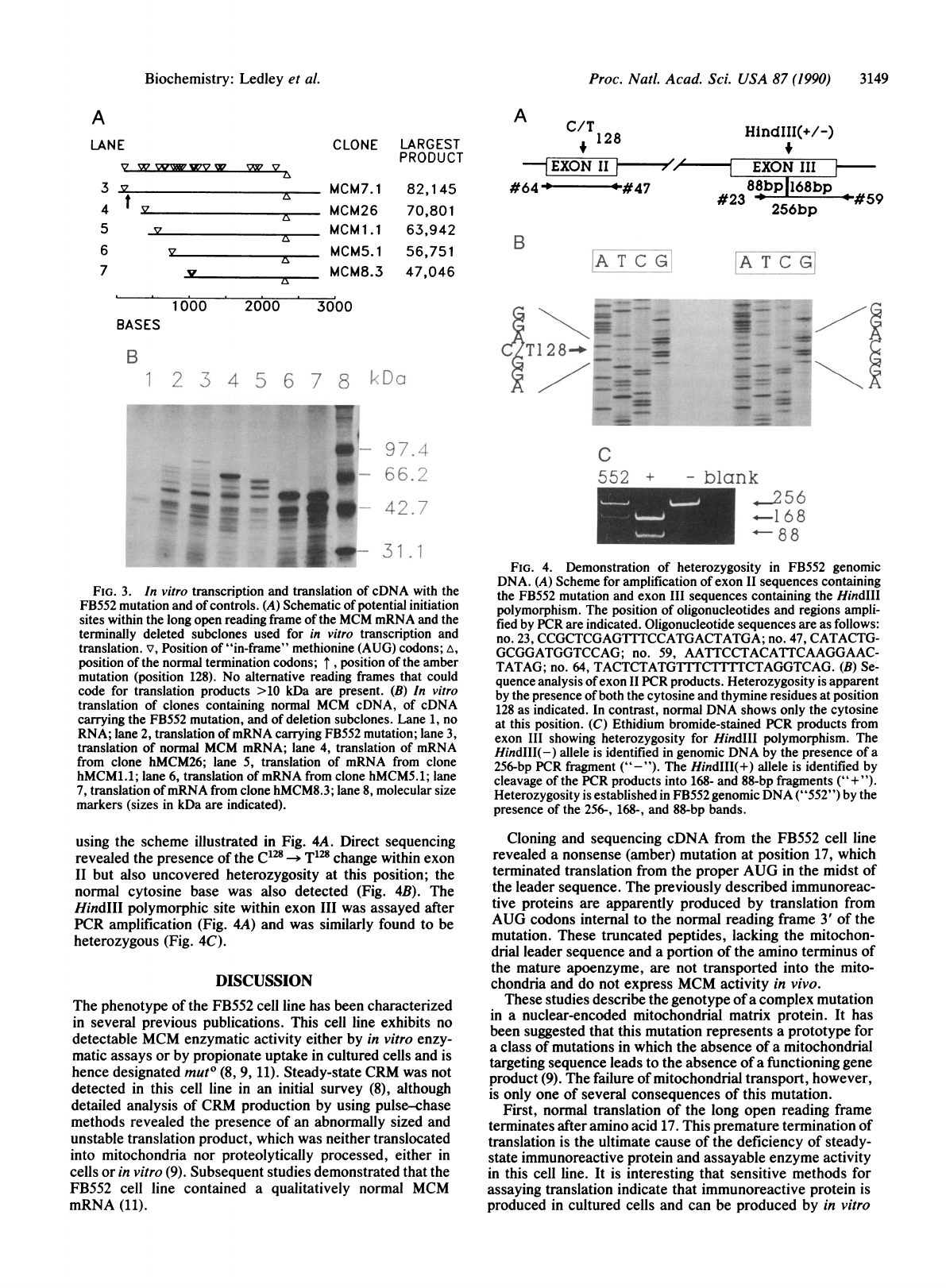
using
the
scheme
illustrated
in
Fig.
4A.
Direct
sequencing
revealed
the
presence
of
the
C128
-*
T128
change
within
exon
II
but
also
uncovered
heterozygosity
at
this
position;
the
normal
cytosine
base
was
also
detected
(Fig.
4B).
The
HindIII
polymorphic
site
within
exon
III
was
assayed
after
PCR
amplification
(Fig.
4A)
and
was
similarly
found
to
be
heterozygous
(Fig.
4C).
DISCUSSION
The
phenotype
of
the
FB552
cell
line
has
been
characterized
in
several
previous
publications.
This
cell
line
exhibits
no
detectable
MCM
enzymatic
activity
either
by
in
vitro
enzy-
matic
assays
or
by
propionate
uptake
in
cultured
cells
and
is
hence
designated
mut0
(8,
9,
11).
Steady-state
CRM
was
not
detected
in
this
cell
line
in
an
initial
survey
(8),
although
detailed
analysis
of
CRM
production
by
using
pulse-chase
methods
revealed
the
presence
of
an
abnormally
sized
and
unstable
translation
product,
which
was
neither
translocated
into
mitochondria
nor
proteolytically
processed,
either
in
cells
or
in
vitro
(9).
Subsequent
studies
demonstrated
that
the
FB552
cell
line
contained
a
qualitatively
normal
MCM
mRNA
(11).
C
k
.
56
--1
68
88
FIG.
4.
Demonstration
of
heterozygosity
in
FB552
genomic
DNA.
(A)
Scheme
for
amplification
of
exon
II
sequences
containing
the
FB552
mutation
and
exon
III
sequences
containing
the
Hind1II
polymorphism.
The
position
of
oligonucleotides
and
regions
ampli-
fied
by
PCR
are
indicated.
Oligonucleotide
sequences
are
as
follows:
no.
23,
CCGCTCGAG1TTCCATGACTATGA;
no.
47,
CATACTG-
GCGGATGGTCCAG;
no.
59,
AATTCCTACATTCAAGGAAC-
TATAG;
no.
64,
TACTCTATGTLTCTITTCTAGGTCAG.
(B)
Se-
quence
analysis
of
exon
II
PCR
products.
Heterozygosity
is
apparent
by
the
presence
of
both
the
cytosine
and
thymine
residues
at
position
128
as
indicated.
In
contrast,
normal
DNA
shows
only
the
cytosine
at
this
position.
(C)
Ethidium
bromide-stained
PCR
products
from
exon
III
showing
heterozygosity
for
HindII1
polymorphism.
The
HindIII(-)
allele
is
identified
in
genomic
DNA
by
the
presence
of
a
256-bp
PCR
fragment
("-").
The
HindIII(+)
allele
is
identified
by
cleavage
of
the
PCR
products
into
168-
and
88-bp
fragments
("+
").
Heterozygosity
is
established
in
FB552
genomic
DNA
("552")
by
the
presence
of
the
256-,
168-,
and
88-bp
bands.
Cloning
and
sequencing
cDNA
from
the
FB552
cell
line
revealed
a
nonsense
(amber)
mutation
at
position
17,
which
terminated
translation
from
the
proper
AUG
in
the
midst
of
the
leader
sequence.
The
previously
described
immunoreac-
tive
proteins
are
apparently
produced
by
translation
from
AUG
codons
internal
to
the
normal
reading
frame
3'
of
the
mutation.
These
truncated
peptides,
lacking
the
mitochon-
drial
leader
sequence
and
a
portion
of
the
amino
terminus
of
the
mature
apoenzyme,
are
not
transported
into
the
mito-
chondria
and
do
not
express
MCM
activity
in
vivo.
These
studies
describe
the
genotype
of
a
complex
mutation
in
a
nuclear-encoded
mitochondrial
matrix
protein.
It
has
been
suggested
that
this
mutation
represents
a
prototype
for
a
class
of
mutations
in
which
the
absence
of
a
mitochondrial
targeting
sequence
leads
to
the
absence
of
a
functioning
gene
product
(9).
The
failure
of
mitochondrial
transport,
however,
is
only
one
of
several
consequences
of
this
mutation.
First,
normal
translation
of
the
long
open
reading
frame
terminates
after
amino
acid
17.
This
premature
termination
of
translation
is
the
ultimate
cause
of
the
deficiency
of
steady-
state
immunoreactive
protein
and
assayable
enzyme
activity
in
this
cell
line.
It
is
interesting
that
sensitive
methods
for
assaying
translation
indicate
that
immunoreactive
protein
is
produced
in
cultured
cells
and
can
be
produced
by
in
vitro
-
WN
.Z
-"W
--*W
Biochemistry:
Ledley
et
al.

Proc.
Natl.
Acad.
Sci.
USA
87
(1990)
methods.
When
the
FB552
cDNA
is
translated
in
vitro,
the
truncated
proteins
appear
to
be
produced
by
coincident
translation
from
internal
AUG
sequences
rather
than
by
reinitiation
of
translation
following
premature
termination,
because
the
smaller
translation
products
are
generated
in
roughly
equivalent
amounts
from
the
normal
and
FB552
cell
lines.
In
contrast,
when
terminally
deleted
subclones
are
used
as
controls,
greater
translational
efficiency
is
seen
from
the
internal
initiation
sites.
These
observations
are
consistent
with
previous
data
indicating
that
the
translational
efficiency
of
mRNA
purified
from
the
FB552
cell
line
is
significantly
lower
than
that
of
other
mRNAs
(9).
Second,
the
translated
products
are
unstable
and
can
only
be
detected
in
cells
by
pulse-chase
or
in
vitro
translation
(9).
There
are
many
potential
explanations
for
this
instability,
including
the
primary
amino-terminal
sequence
(19)
or
ter-
tiary
configuration
of
the
truncated
peptide
or
a
failure
of
proper
compartmentalization
(9,
20).
The
present
work
does
not
address
the
mechanism
underlying
this
instability.
The
impaired
translation
of
the
long
open
reading
frame
distal
to
the
amber
mutation
and
the
relative
instability
of
this
abnor-
mal
protein
product
constitute
the
proximal
cause
of
the
absence
of
steady-state
protein
and
assayable
enzyme
activ-
ity.
Third,
in
the
absence
of
a
leader
sequence,
the
truncated
translation
products
are
not
transported
into
mitochondria.
Even
if
these
proteins
retained
determinants
for
necessary
catalytic
activity,
it
is
doubtful
that
this
gene
product
would
be
biologically
active
in
the
cytoplasmic
space.
The
adeno-
sylcobalamin
cofactor,
required
for
holoenzyme
formation
and
activity,
is
synthesized
within
the
mitochondria
and
is
not
transported
into
the
cytoplasm.
In
addition,
the
methyl-
malonyl-CoA
substrate
is
also
synthesized
within
the
mito-
chondria
(5).
In
addition,
protein
factors
responsible
for
proper
assembly
of
mitochondrial
proteins
are
not
present
in
the
cytosol
(15).
Finally,
although
only
one
cDNA
species
was
cloned
from
this
cell
line,
analysis
of
genomic
DNA
indicates
that
this
cell
line
is
heterozygous
for
both
the
C128
mutation
and
a
poly-
morphic
HindIII
site.
It
was
previously
suggested
that
the
multiple
bands
of
immunoreactive
protein
observed
in
the
FB552
cell
line
represented
translation
products
of
mRNA
species
arising
from
heterozygous
alleles.
Analysis
of
poly-
morphisms
within
the
MUT
genomic
locus
indicates
that
this
cell
line
is
indeed
a
compound
heterozygote.
One
allele
contains
the
C128
-*
T128
mutation.
The
second
allele,
how-
ever,
apparently
does
not
express
a
clonable
mRNA
because
pools
of
amplified
cDNA
contain
only
T'28/HindIII(-)
tran-
scripts.
Thus,
it
is
likely
that
the
multiple
translation
products
observed
previously
arose
from
coincident
usage
of
alterna-
tive
AUG
codons
within
the
mutant
mRNA,
as
seen
in
the
in
vitro
translation
experiment,
rather
than
from
translation
from
heterologous
alleles.
Previous
studies
have
shown
that
up
to
one-third
of
mut°
cell
lines
express
a
"low
message"
phenotype
in
which
the
levels
of
hybridizable
mRNA
are
significantly
lower
than
in
normal
cell
lines
(11).
This
observation
is
consistent
with
protein
data,
which
predicted
the
presence
of
"null"
alleles
among
the
CRM+
cell
lines
(9).
It
is
likely
that
these
low
message
alleles
are
common
contributors
to
compound
het-
erozygosity
in
both
mut-
and
mut°
forms
of
MMA.
The
nature
of
this
mutation
(or
mutations)
in
genomic
DNA
has
yet
to
be
determined.
This
work
represents
an
application
of
molecular
cloning
to
the
characterization
of
mutations
in
mut
MMA.
These
studies
confirm
the
predictions
made
by
somatic
cell
complementa-
tion
and
analysis
of
the
biochemical
phenotype
of
MMA
cells
that
the
mut
complementation
group
represents
cells
with
allelic
mutations
within
the
MUT
locus.
A
variety
of
muta-
tions
have
now
been
described,
which
underlie
mut
MMA
(R.J.,
S.-U.N.,
and
F.D.L.,
unpublished
results).
There
is
no
evidence
from
population
genetic
data
or
molecular
cloning
that
MMA
represents
a
discrete
set
of
common
mutations.
Rather,
this
relatively
rare
disorder
appears
to
result
from
a
large
variety
of
mutations
as
would
be
expected
if
a
genetic
equilibrium
exists
between
new
mutations
and
the
generally
lethal
nature
of
MCM
deficiency.
The
authors
gratefully
acknowledge
the
contributions
of
Ana
Maria
Crane
and
Adelle
Hack
in
characterizing
the
FB552
cell
line
and
Tammy
Reid
in
preparing
this
manuscript.
This
work
was
supported
by
National
Institutes
of
Health
Grants
HD-24186
(F.D.L.)
and
DK-12579
(L.E.R.)
and
the
Mental
Retardation
Re-
search
Center
at
the
Baylor
College
of
Medicine
(P30-HD-24064
to
Dr.
Ed
McCabe).
F.D.L.
is
an
Assistant
Investigator
of
the
Howard
Hughes
Medical
Institute.
1.
Kolhouse,
J.
F.,
Utley,
C.
&
Allen,
R.
H.
(1980)
J.
Biol.
Chem.
255,
2708-2712.
2.
Fenton,
W.
A.,
Hack,
A.
M.,
Willard,
H.
F.,
Gertler,
A.
&
Rosenberg,
L.
E.
(1982)
Arch.
Biochem.
Biophys.
214,
815-
823.
3.
Fenton,
W.
A.,
Hack,
A.
M.,
Helfgott,
D.
&
Rosenberg,
L.
E.
(1984)
J.
Biol.
Chem.
259,
1-21.
4.
Jansen,
R.,
Kalousek,
F.,
Fenton,
W.
A.,
Rosenberg,
L. E.
&
Ledley,
F.
D.
(1989)
Genomics
4,
198-205.
5.
Rosenberg,
L.
E.
&
Fenton,
W.
A.
(1989)
in
The
Metabolic
Basis
of
Inherited
Disease,
eds.
Scriver,
C.
R.,
Beaudet,
A.
L.,
Sly,
W.
S.
&
Valle,
D.
(McGraw-Hill,
New
York),
6th
Ed.,
pp.
822-844.
6.
McKusick,
V.
A.
(1988)
Mendelian
Inheritance
in
Man
(Johns
Hopkins
Univ.
Press,
Baltimore).
7.
Willard,
H.
F.
&
Rosenberg,
L.
E.
(1980)
J.
Clin.
Invest.
5,
90-98.
8.
Kolhouse,
J.
F.,
Utley,
C.,
Fenton,
W.
A.
&
Rosenberg,
L.
E.
(1981)
Proc.
Natd.
Acad.
Sci.
USA
78,
7737-7741.
9.
Fenton,
W.
A.,
Hack,
A.
M.,
Kraus,
J.
P.
&
Rosenberg,
L.
E.
(1987)
Proc.
Nati.
Acad.
Sci.
USA
84,
1421-1424.
10.
Ledley,
F.
D.,
Lumetta,
M.,
Nguyen,
P.
N.,
Kolhouse,
J.
F.
&
Allen,
R.
A.
(1988)
Proc.
Natl.
Acad.
Sci.
USA
85,
3518-3521.
11.
Ledley,
F.
D.,
Crane,
A.
M.
&
Lumetta,
M.,
Am.
J.
Hum.
Genet.,
in
press.
12.
Vrieling,
H.,
Simons,
J.
W.
I.
M.
&
vanZeeland,
A.
A.
(1988)
Mutat.
Res.
198,
107-113.
13.
Scherrer,
K.
(1969)
in
Fundamental
Techniques
in
Virology,
eds.
Habel,
K.
&
Salzman,
N.
P.
(Academic,
New
York),
pp.
413-432.
14.
Berger,
S.
B.
&
Kimmel,
A.
R.
(1987)
Guide
to
Molecular
Cloning
Techniques
(Academic,
New
York).
15.
Cheng,
M.
Y.,
Hartl,
F.
U.,
Martin,
J.,
Pollock,
R.
A.,
Kalousek,
F.,
Neupert,
W.,
Hallberg,
E.
M.,
Hallberg,
R.
L.
&
Horwich,
A.
L.
(1989)
Nature
(London)
337,
620-625.
16.
Scarf,
S.
J.,
Horn,
G.
T.
&
Erlich,
H.
A.
(1986)
Science
233,
1076-1078.
17.
Gibbs,
R.
A.,
Nguyen,
P.
N.,
McBride,
L.
J.,
Koepf,
S.
M.
&
Caskey,
C.
T.
(1989)
Proc.
Natl.
Acad.
Sci.
USA
86,
1919-
1923.
18.
van
der
Werf,
S.,
Bradely,
J.,
Wimer,
E.,
Studier,
F.
W.
&
Dunn,
J.
J.
(1986)
Proc.
Natl.
Acad.
Sci.
USA
83,
2330-2334.
19.
Bachmair,
A.
&
Varshavsky,
A.
(1989)
Cell
56,
1019.
20.
Horwich,
A.
L.,
Fenton,
W.
A.,
Figaira,
F.
A.,
Fox,
J.
E.,
Kolansky,
D.,
Mellman,
I.
S.
&
Rosenberg,
L.
E.
(1985)
J.
Cell
Biol.
100,
1515-1521.
3150
Biochemistry:
Ledley
et
al.
