
STRENGTHENING
OUR NATIONAL SYSTEM
FOR MEDICAL DEVICE
POSTMARKET
SURVEILLANCE
CENTER FOR DEVICES AND RADIOLOGICAL HEALTH
U.S. FOOD AND DRUG ADMINISTRATION
APRIL 2013
UPDATE AND NEXT STEPS

02STRENGTHENING OUR NATIONAL SYSTEM FOR MEDICAL DEVICE POSTMARKET SURVEILLANCE: UPDATE AND NEXT STEPS
In September 2012, the Food and Drug
Administration's (FDA), Center for De-
vices and Radiological Health (CDRH)
issued a report entitled "Strengthening
Our National System for Medical Device
Postmarket Surveillance."
1
FDA's vision
for medical device postmarket surveil-
lance is the creation of a national sys-
tem that serves four primary functions:
Communicates timely, accurate, sys-
tematic, and prioritized assessments
devices throughout their marketed life
using high quality, standardized, struc-
tured, electronic health-related data;
in near real-time from a variety of
privacy-protected data sources;
Reduces the burdens and costs of
medical device postmarket surveil-
lance; and
Facilitates the clearance and approval
of new devices, or new uses of exist-
ing devices.
The report contained four key proposed
National System:
(UDI) system and promote its in-
corporation into electronic health
information;
2. Promote the development of national
and international device registries for
selected products;
3. Modernize adverse event reporting
and analysis; and
4. Develop and use new methods for
evidence generation, synthesis, and
appraisal.
The FDA recognizes that input and ac-
tive participation from many key national
and international stakeholders is necessary
to strengthen medical device postmarket
surveillance and that a national system
cannot be implemented or achieved by the
FDA alone. Therefore, following release
of the report, FDA held a series of public
meetings in September 2012 and accepted
comments via our website to garner stake-
holder feedback.
2
This update incorporates the public input
we received and describes the next steps
FDA intends to take to establish a Na-
tional Medical Device Postmarket Sur-
veillance System.
BACKGROUND
1
Report available at: http://www.fda.gov/AboutFDA/CentersOces/OceofMedicalProductsandTobacco/CDRH/CDRHReports
UCM301912
2
http://www.fda.gov/MedicalDevices/NewsEvents/ WorkshopsConferences/ucm111051.htm

03STRENGTHENING OUR NATIONAL SYSTEM FOR MEDICAL DEVICE POSTMARKET SURVEILLANCE: UPDATE AND NEXT STEPS
Medical device postmarket surveillance
presents unique challenges compared to
drugs and biologics due to the greater
diversity and complexity of medical
devices, the iterative nature of medical
product development, the learning curve
associated with technology adoption, and
the relatively short product life cycle.
The FDA believes that privacy-protected,
routinely collected electronic health
information containing UDI and device-
3
in selected product areas
complemented by additional data sources
(e.g. adverse event reports, administrative
and claims data) should serve as the
foundation for such a national medical
device postmarket surveillance system
(Figure).
Such a system would have broad patient
capture, real-world generalizability, scalable
and reusable infrastructure, continuous
accrual of information for near real-time
analysis, and use structured data with
It would be capable of providing near
risks of medical devices throughout their
marketed life, identifying potential safety
signals, reducing the burdens and costs of
medical device postmarket surveillance,
and facilitating the clearance and approval
of new devices, or new uses of existing
devices.
In 2012, the FDA issued the proposed rule
for a UDI system for all medical devices
4
,
based approach to UDI implementation,
devices and exempting lower risk devices
from some or all of the requirements.
surveillance activities by providing a stan-
dard and unambiguous way to document
device use in electronic health records,
clinical information systems, claims data
sources, and registries, potentially making
vast amounts of previously untapped clin-
ical information available for assessing the
-
ing data sources (like registries and claims
data).
Registries play a unique and prominent
role in medical device surveillance because
they can provide additional detailed infor-
mation about patients, procedures, and
devices not routinely collected by electron-
ic health records, administrative or claims
data. For this reason, registries will contin-
ue to serve a critical, complementary role
in medical device postmarket surveillance,
even as UDI becomes more routinely in-
corporated into electronic health informa-
tion. The creation of individual registries
-
LAYING THE FOUNDATION FOR AN INTEGRATED SYSTEM
3
A registry is a system that collects and maintains structured records on a specic disease, condition, procedure, or
medical product for a specied time period and population.
4
http://www.fda.gov/medicaldevices/deviceregulation andguidance/uniquedeviceidentication/default.htm
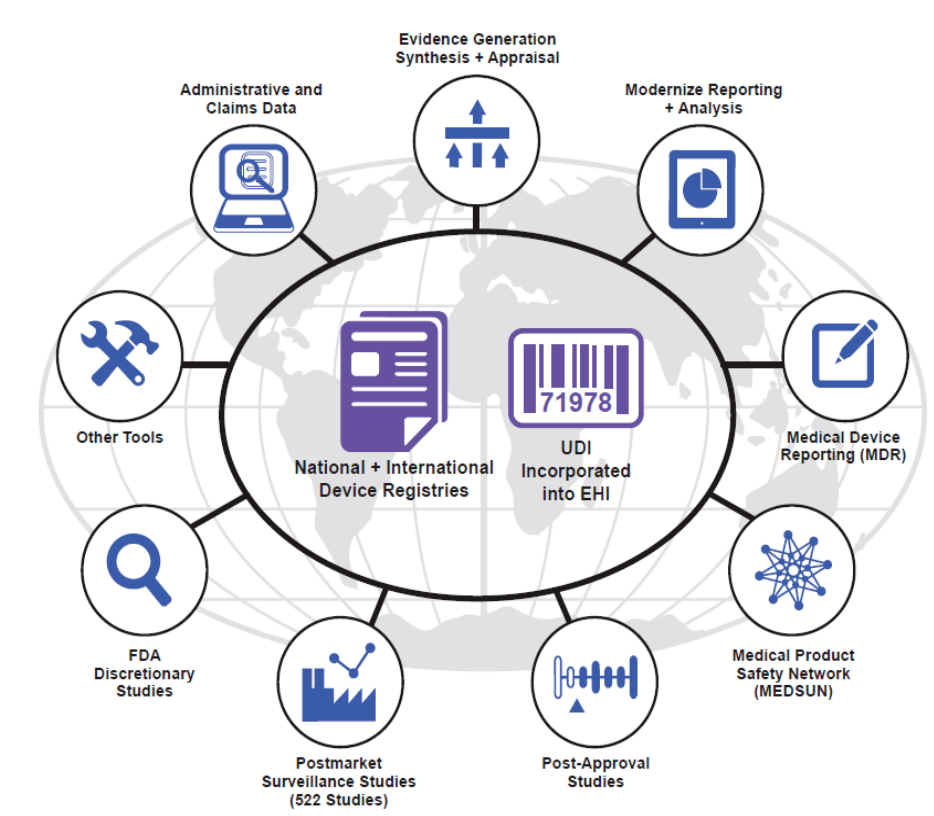
04STRENGTHENING OUR NATIONAL SYSTEM FOR MEDICAL DEVICE POSTMARKET SURVEILLANCE: UPDATE AND NEXT STEPS
Figure. FDA believes that national and international device registries in selected product areas
the foundation of our National Medical Device Postmarket Surveillance System. The system
could be linked to longitudinal data, such as administrative and claims data, and would employ
novel methods for evidence generation, synthesis and appraisal, modernized adverse event
reporting and analysis, and would complement existing tools such as Medical Device Reporting
(MDR), an enhanced surveillance network of approximately 280 hospitals (Medical Product
Safety Network – MedSun), studies ordered by the FDA for selected devices (Post-Approval
Studies and Postmarket Surveillance Studies), FDA research using other data sources (FDA
Discretionary Studies), and other tools such as device tracking.

05STRENGTHENING OUR NATIONAL SYSTEM FOR MEDICAL DEVICE POSTMARKET SURVEILLANCE: UPDATE AND NEXT STEPS
it is impractical and unnecessary to have
registries for every medical device type.
Instead, targeted registry efforts should
be based on wide stakeholder input and
support and should focus on selected ar-
large public health need, patient exposure,
uncertain long-term or real-world device
performance, or societal cost.
The Food and Drug Administration Safe-
ty and Innovation Act of 2012 (FDASIA)
requires expansion of FDA’s Sentinel
System
5
to include medical devices. The
current Sentinel data model focuses on
querying privacy-protected administrative
and claims data complemented, in part, by
information in electronic health records
(such as lab results) and maintained by
partner organizations. Unfortunately, most
records accessible to Sentinel lack manu-
-
leveraged to perform meaningful medical
device postmarket surveillance. However,
as UDIs are implemented and adopted
throughout the healthcare system, current
efforts can be expanded to include this
essential information for the purpose of
medical device postmarket surveillance.
Ultimately, we envision a medical device
postmarket surveillance system using
distributed data sources, focusing on
electronic health information containing
UDI and registries linked to, or integrated
with, other longitudinal data sources. FDA
is not seeking to develop a centralized
repository of medical device-related
electronic health information. Rather each
data owner should retain physical and
operational control over their data, provide
input into valid use and data interpretation,
and maintain patient privacy.
5
http://www.fda.gov/Safety/FDAsSentinelInitiative/default.htm

06STRENGTHENING OUR NATIONAL SYSTEM FOR MEDICAL DEVICE POSTMARKET SURVEILLANCE: UPDATE AND NEXT STEPS
In 2013, the FDA intends to pursue the
following critical efforts towards creating
a national medical device postmarket
surveillance system (see Appendix):
1. Establish a multi-stakeholder Medi-
cal Device Postmarket Surveillance
System Planning Board
6
to identify
the governance structure, practices,
policies, procedures, methods and
business model(s) necessary to fa-
cilitate the creation of a sustainable,
integrated medical device postmarket
surveillance system that leverages and
complements existing and on-going
efforts.
2. -
tion (UDI) system and promote its
incorporation into electronic health
information.
functional and publically accessible
global UDI database (GUDID) to
device information to stakeholders
and the general public.
Take steps to facilitate the incor-
poration of UDI into electronic
health records as part of EHR
Complete a pilot demonstrating the
ability to incorporate UDI into a
multi-hospital information system.
Complete an initial think
tank report to inform the
development of a roadmap for
successful UDI implementation.
7
3. Promote the development of national
and international device registries for
selected products.
Establish a Medical Device Registry
Task Force consisting of key reg-
istry stakeholders under CDRH’s
Medical Device Epidemiology Net-
work (MDEpiNet)
8
Program.
IMPLEMENTATION
6
FDA intends to issue a public call for nominations to establish a multi-stakeholder Planning Board that includes the medical
device industry, health care provider community, medical professional societies, patient and consumer groups. third-party
payers, hospitals and other health care facilities, health care data centers, government agencies, and other relevant stakeholders.
The public will have an opportunity to comment on recommendations from the Planning Board before a decision is made to
implement them.
7
This eort is conducted in collaboration with the Engelberg Center for Health Care Reform at Brookings and Chickasaw Nation
Industries, Inc. See www.brookings.edu/about/centers/health/projects/development-and-use-of-medical-devices.
8
http://www.fda.gov/MedicalDevices/ScienceandResearch/EpidemiologyMedicalDevices/
MedicalDeviceEpidemiologyNetworkMDEpiNet/default.htm

07STRENGTHENING OUR NATIONAL SYSTEM FOR MEDICAL DEVICE POSTMARKET SURVEILLANCE: UPDATE AND NEXT STEPS
4. Modernize adverse event reporting
and analysis.
†
pilots to detect and automatically
report to FDA select device-related
adverse events though hospital
electronic health records and
incident reporting systems.
Implement a mobile application for
voluntary adverse event reporting.
Pilot an initial functional release of
the FDA Adverse Event Reporting
System (FAERS), a modernized
database for adverse event reports.
Implement prospective “data min-
ing” tools in at least three major
device areas to enhance the iden-
event reports and report trends and
clusters.
5. Develop and use new methods for
evidence generation, synthesis, and
appraisal.
Identify gaps in current method-
ological efforts to promote data sta
ndardization, interoperability, and
linkage between registries and dispa-
rate data sources.
Advance the development of in-
teroperability between registries and
electronic health records.
Collaborate with stakeholders and
leverage on-going efforts to develop
an approach for evidence synthesis
to integrate registry-based informa-
tion with other data sources to pro-
vide more timely and comprehensive
†
ADE Spontaneous Triggered Electronic Reporting for Devices
08STRENGTHENING OUR NATIONAL SYSTEM FOR MEDICAL DEVICE POSTMARKET SURVEILLANCE: UPDATE AND NEXT STEPS
CONCLUSION
Postmarket surveillance of medical devices presents unique challenges. Although the Unit-
ed States has a robust medical device postmarket surveillance system, we believe it can be
strengthened by developing a more integrated national system. Our planned strategic actions
will complement our existing programs and other ongoing efforts and can be accomplished
under our existing authorities within our current budget. These efforts, and the envisioned
medical device postmarket surveillance system as a whole, are intended to be collaborative
and transparent because we recognize that our postmarket vision cannot be implemented or
achieved by the FDA alone.
We will continue to provide updates on our postmarket activities on our website.
09STRENGTHENING OUR NATIONAL SYSTEM FOR MEDICAL DEVICE POSTMARKET SURVEILLANCE: UPDATE AND NEXT STEPS
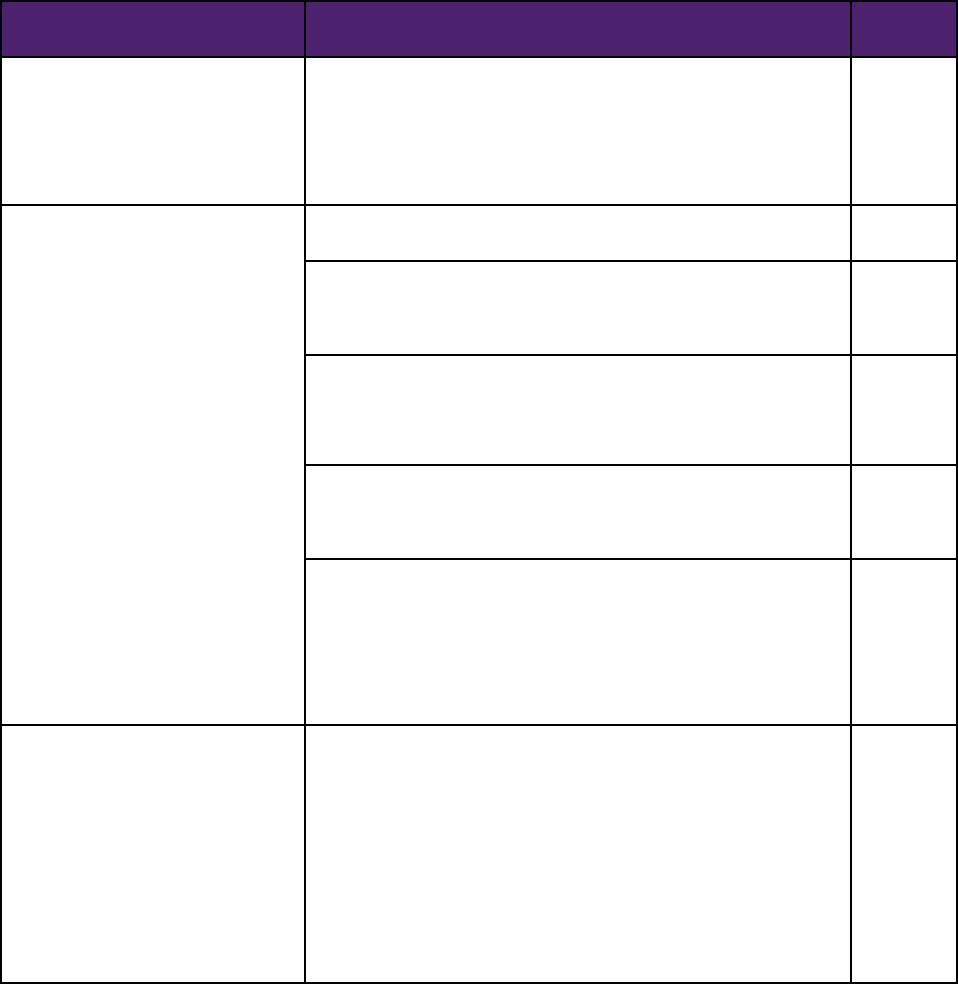
ITEM
ACTION
Deliverable
Date
Medical Device Postmarket
Surveillance System Governance
Establish a multi-stakeholder medical device postmarket surveillance
system planning board to identify the governance structure, practices,
policies, procedures, methods and business model(s) necessary to
facilitate the creation of a sustainable, integrated medical device
postmarket surveillance system that leverages and complements existing
and on-going efforts.
9/30/2013
Establish a unique device
identification (UDI) system and
promote its incorporation into
electronic health information
Finalize the Unique Device Identification (UDI) rule.
6/30/2013
Develop and implement a fully functional and publically accessible global
UDI database (GUDID) to provide detailed, non-confidential device
information to stakeholders and the general public.
6/30/2013
Provide UDI technical requirements and UDI electronic health record
(EHR) use cases to Office of the National Coordinator for Health
Information Technology (ONC) standards workgroups to facilitate UDI
adoption as part of EHR certification.
9/30/2013
Complete technical and final reports on a pilot, demonstrating issues and
challenges involved in incorporating UDI into a multi-hospital information
system.
12/31/2013
Complete an initial thinktank report to inform the development of a
roadmap for successful UDI implementation addressing critical issues
including: 1) opportunities and challenges associated with capturing UDIs
in claims; 2) steps necessary for implementation and integration of UDI
within electronic data infrastructure of care delivery sites; and 3) patient
and provider access to and linking of device information across data
sources.
6/30/2013
Promote the development of national
and international device registries
for selected products
Establish a Medical Device Registry Task Force consisting of key
stakeholders under CDRH’s Medical Device Epidemiology Network
(MDEpiNet) Program to: 1) identify existing registries that may contribute
to the system; 2) leverage on-going registry efforts focused on quality
improvement, reimbursement, patient-centered outcomes and other
activities to best meet the needs of multiple stakeholders; 3) identify
priority medical device types for which the establishment of a longitudinal
registry is of significant public health importance; 4) define registry
governance and data quality practices that promote rigorous design,
conduct, analysis, and transparency to meet stakeholder needs; and 5)
develop strategies for the use of registries to support premarket approval
and clearance.
6/30/2013
APPENDIX
STRENGTHENING OUR NATIONAL SYSTEM FOR MEDICAL DEVICE POSTMARKET SURVEILLANCE
Update and Next Steps
SUMMARY OF
2013 PLANNED FDA ACTION
10STRENGTHENING OUR NATIONAL SYSTEM FOR MEDICAL DEVICE POSTMARKET SURVEILLANCE: UPDATE AND NEXT STEPS
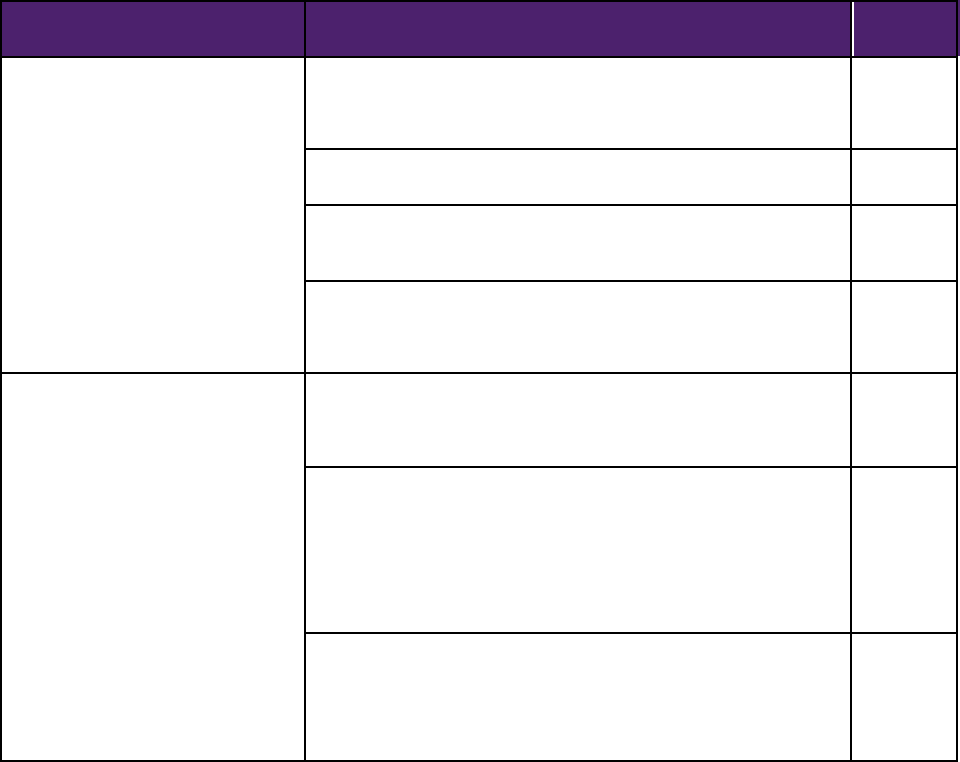
ITEM
ACTION
Deliverable
Date
Modernize adverse event reporting
and analysis
Issue final reports on two pilots demonstrating use of ASTER-D for the
detection and automated reporting of select device-related adverse
events though hospital electronic health records.
12/31/2013
Implement a mobile application for voluntary adverse event reporting.
6/30/2013
Pilot an initial functional release of the FDA Adverse Event Reporting
System (FAERS), a modernized database for adverse event reports.
12/31/2013
Implement prospective “data mining” tools in at least three major device
areas to enhance the identification of high quality adverse event reports
and report trends and clusters.
9/30/2013
Develop and use new methods for
evidence generation, synthesis and
appraisal
Identify gaps in current methodological efforts to promote data
standardization, interoperability, and linkage between registries and
disparate data sources.
9/30/2013
Advance the development of interoperability between registries and
electronic health records by:
1) beginning collaboration with stakeholders on the use of structured data
capture capabilities within electronic health information sources; 2)
facilitating a demonstration pilot focusing on registry-EHR integration; and
3) assessing methodologies to optimize linkage of registries with other
longitudinal data sources such as administrative and claims data.
9/30/2013
Collaborate with stakeholders and leverage on-going efforts to develop
methodology for evidence synthesis for two classes of implantable
devices to combine diverse data sources and/or combine data from
multiple registries to provide more timely and comprehensive
assessments of device benefit-risk profiles.
9/30/2013
STRENGTHENING OUR NATIONAL SYSTEM FOR MEDICAL DEVICE POSTMARKET SURVEILLANCE
Update and Next Steps
SUMMARY OF
2013 PLANNED FDA ACTION, Continued

Food and Drug Administration
Center for Devices and Radiological Health
10903 New Hampshire Avenue
Silver Spring, MD 20993
http://www.fda.gov/MedicalDevices
http://www.fda.gov/Radiation-EmittingProducts
